JGI portals use the NCBI's BLAST (Basic Local Alignment Search Tool algorithm and Kent Informatics' BLAT (BLAST-Like Alignment Tool) to allow users to identify regions of nucleotide/protein similarity between biological organisms and perform rapid mRNA/DNA and cross-species protein alignments between a sequence of their choosing and the annotated genomes in the portal databases.
BLAST/BLAT searches are conducted ONLY against the selected portal database, not the NCBI/UCSC databases.
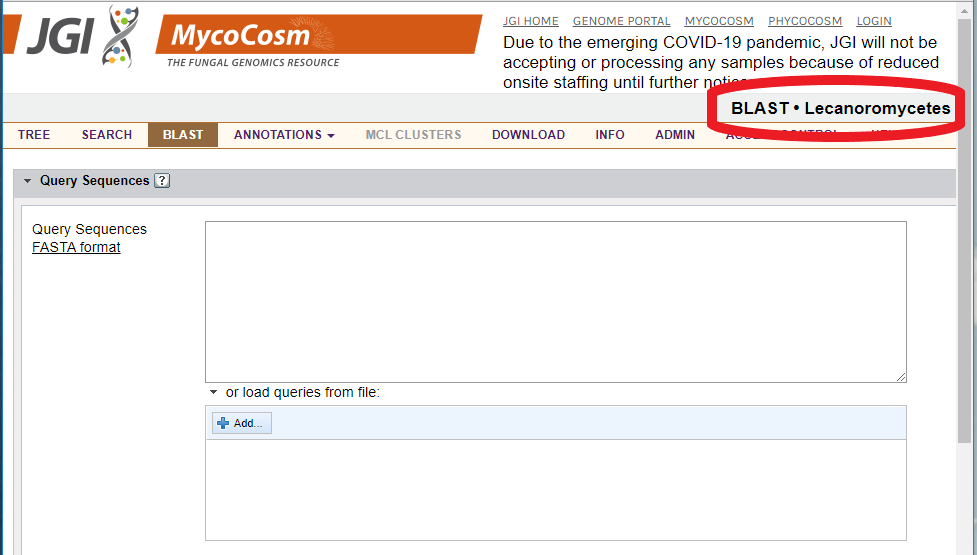
BLAST/BLAT searches are context dependent. You will be comparing your unknown sequence only to those genomes indicated in the upper right-hand corner of the screen:
In this example, you will be comparing your nucleotide or protein sequence to the genomes in the Mycocosm database from the class Lecanoromycetes only.
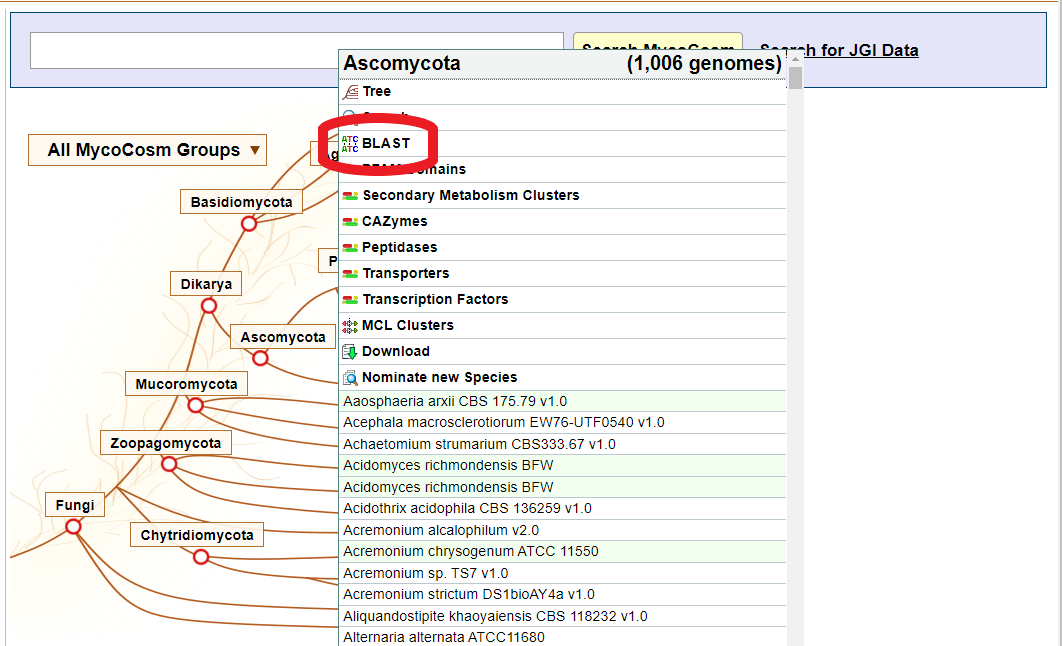 If you want to broaden your search and compare your sequence to all the genomes in the phylum Ascomycota, simply return to the Home Page, select Ascomycota, and then select the BLAST link.
If you want to broaden your search and compare your sequence to all the genomes in the phylum Ascomycota, simply return to the Home Page, select Ascomycota, and then select the BLAST link.
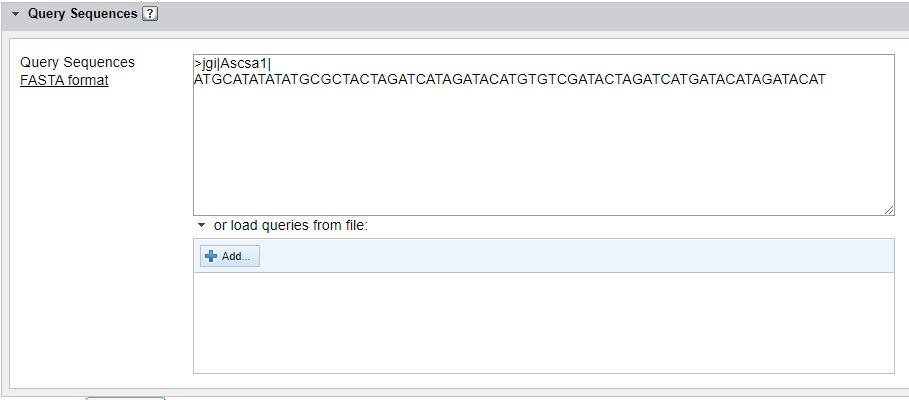 Nucleotide or amino acid sequences can be entered directly into the empty field or uploaded using the +ADD button. Sequences must be in FASTA format. For more information on formatting, please click here:FASTA Formatting Guidelines)
Nucleotide or amino acid sequences can be entered directly into the empty field or uploaded using the +ADD button. Sequences must be in FASTA format. For more information on formatting, please click here:FASTA Formatting Guidelines)
Your alignment run will search against BOTH sequences directly into the query box and sequences included in any uploaded FASTA files.
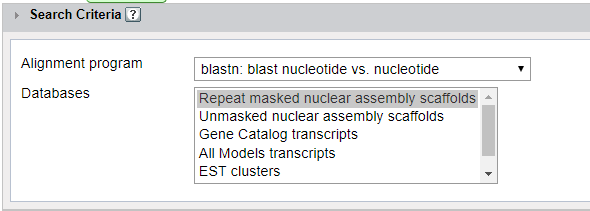
Once your sequences have been entered, you will need to select the alignment program you wish to use and the database you would like to search against.
Alignment searches may be run using a variety of programs which compare DNA, protein, or translated nucleotide sequences. These programs are based on three families of alignment algorithms, NCBI's BLAST), Webb Miller's Blastz), and Jim Kent's BLAT (BLAST-Like Alignment Tool). Please see the Ensemble Help Page for the differences between BLAST and BLAT. Briefly, BLAT and BLAST are similar however BLAT requires a near perfect match to generate a hit and is quicker at identifying closely related sequences however BLAST is more flexible and is capable of finding more remote matches.
tblastx: For identifying nucleotide sequences similar to the query based on their coding potential tblastn: For identifying database sequences encoding proteins similar to the query blastn: General purpose nucleotide search and alignment program that is sensitive and can be used to align tRNA or rRNA sequences as well as mRNA or genomic DNA sequences containing a mix of coding and noncoding regions blastx: Compares the six-frame conceptual translation products of a nucleotide query sequence (both strands) against a protein sequence database blastp: Used for both identifying a query amino acid sequence and for finding similar sequences in protein databases tblatn: Same as tblastn but using the BLAT algorithm tblatx: Same as tblastx but using the BLAT algorithm blatn: Same as blastn but using the BLAT algorithm blatp: Same as blastp but using the BLAT algorithm blatx: Same as blastx but using the BLAT algorithm
Repeat masked nuclear assembly scaffolds: This is the soft-masked genome assembly. Generally, this should for tblastn and blastn against the genomic sequence. Unmasked nuclear assembly scaffolds: This is the unmasked genome sequence. This can be used when looking for repeats, TEs, transposons, etc. This is also a good choice when looking for small genes that might otherwise be missed. Gene Catalog transcripts: This is the transcripts from the predicted genes in the genome (one gene per locus). All Models transcripts: This has transcripts from all gene predictors used in the annotation pipeline; multiple models per locus. EST clusters: Search clusters of expressed sequence tags ESTs: Search expressed sequence tags. An expressed sequence tag is a short sub-sequence of a cDNA sequence. ESTs may be used to identify gene transcripts, and are instrumental in gene discovery and in gene-sequence determination. Gene Catalog Proteins: This are the protein sequences of the predicted genes in the genome (one gene per locus). All Model Proteins: This is all protein sequences from all the gene predictors used in the annotation pipeline (many genes per locus).
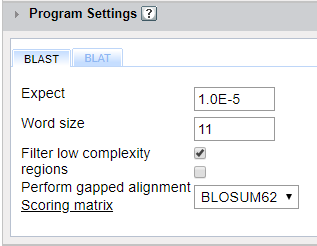
In the program panel, you can set values for the parameters used by BLAST and BLAT alignment programs.
Expect (Default = 1.0E-5) The Expect value (E) is a parameter that describes the number of hits one can "expect" to see by chance when searching a database of a particular size. It decreases exponentially as the Score (S) of the match increases. The lower the E-value, or the closer it is to zero, the more "significant" the match is. The Expect value can be used as a convenient way to create a significance threshold for reporting results. You can change the Expect value threshold on most BLAST search pages. When the Expect value is increased from the default value of 10, a larger list with more low-scoring hits can be reported.
Word Size (Default = 3) The BLAST algorithm uses "words" to nucleate regions of similarity. Changing the initial word-size can help to find more, but less accurate hits; or to limit the results to almost perfect hits. Decreasing the word-size will increase the number of detected homologous sequences, but hits can include alignments of higher fragmentation due to gaps and substitutions. Increasing the word-size will give less hits as it requires a longer continuous regions of exact match. If the word-size is chosen to be almost the size of the query, BLAST will search for almost exact matches (example: search for location of gene sequences in the original genome of the gene) For short sequences, word-size must be less than half the query length, otherwise reliable hits can be missed.
Filter Low Complexity Regions (Default = selected): In BLAST searches performed without this filter selected, high scoring hits may be reported only because of the presence of a low-complexity region. Most often, it is inappropriate to consider this type of match as the result of shared homology. Rather, it is as if the low-complexity region is "sticky" and is pulling out many sequences that are not truly related.
Perform Gapped Alignment: Checking this box allows the BLAST program to add gaps to the alignment. Gaps suggest evolutionary distance and gap penalties are used to reduce the score. Using gapped alignment may allow you to increase the local alignment score despite the inclusion of these penalties.
Scoring Matrix: A scoring matrix is designed to identify distant evolutionary relationships. Empirical replacement frequency scoring matrices can be divided into two types: those extrapolating from an explicit evolutionary model (PAM series) and those calculated from local alignments (BLOSUM -BLOcks SUbstitution Matrix- scoring matrices). You can choose from 2 PAM and 3 BLOSUM matrix options here.
BLOSUM62- Matrix built using sequences with less than 62% similarity. BLOSUM 62 is the default matrix for protein BLAST. Experimentation has shown that the BLOSUM-62 matrix is among the best for detecting most weak protein similarities. PAM30- Model based matrix corresponding to 75% identity PAM70- Model based matrix corresponding to 55% identity BLOSUM45- Designed to compare distantly related alignments BLOSUM80- Designed to compare closely related alignments
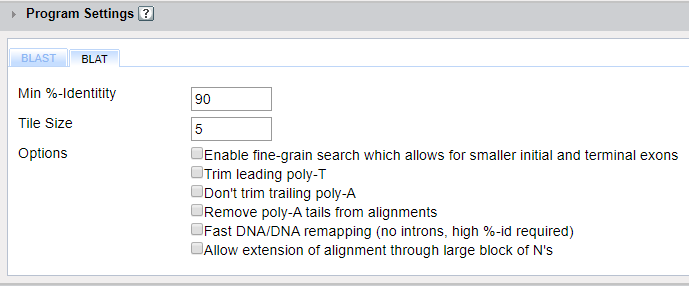
For additional information on BLAT, **CLICK HERE**
Min %-identity- In this field you can define the minimum percent identity that will be shown in the alignment results.
Tile Size- The first step in a BLAT alignment is to split the reference sequence into “tiles”. The size of those tiles is specified here and the default value depends on the alignment program selected. Changing the tile size can change the sensitivity of the program and may slow the speed and increase memory requirements.
The following BLAT search options are available and allow for customization of your search parameters.
Enable fine-grain search which allows for smaller initial and terminal exons: Trim leading Poly-T Don’t trim leading Poly-A Remove poly-A tails from alignment Fast DNA/DNA remapping (no introns, high %-id required) Allow extension of alignment through large blocks of N’s
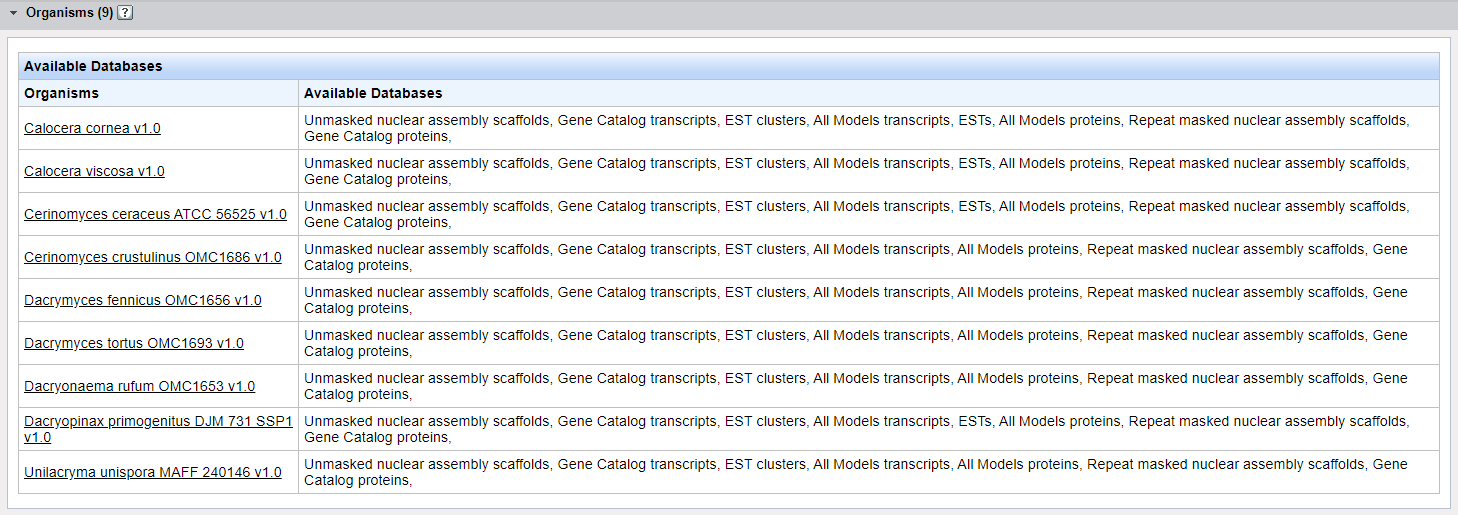 The Organisms Panel appears when you are running an alignment search across an organism group and displays the list of genomes to be included in the search and lists the databases available for that organism. Click on any genome within this list and you will be taken to the corresponding JGI Organism Portal.
The Organisms Panel appears when you are running an alignment search across an organism group and displays the list of genomes to be included in the search and lists the databases available for that organism. Click on any genome within this list and you will be taken to the corresponding JGI Organism Portal.